

货号:D1224
存储条件:-20℃,有效期2年
产品组分
组分 | 规格 | 规格 |
Golden Gate Mix (BsmBI) | 10ul | 50ul |
10× T4 DNA Ligase Buffer | 1ml | 1ml |
产品说明
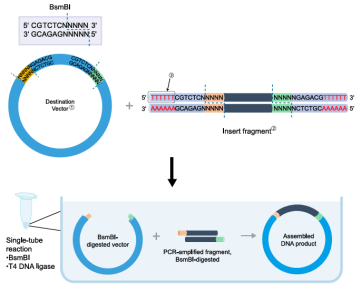
Golden Gate Assembly Kit为系列产品,包括几种不同TypeIIS型限制酶,本产品采用的限制酶为BsmBl。该系列产品均基于Golden Gate Assembly原理,即通过Type IlS 限制酶独特的切割特点得到想要的的粘性末端并通过 T4 DNA Ligase 对其进行连接。此方法尤其擅长组装难以克隆的序列,如重复序列、高GC序列、TAL(transcription activator-like)效应基因、超短序列(< 100 bp)等。Type IIS 型限制性内切酶与传统的限制酶不同,它识别非回文序列并在距其识别位点下游一定距离的位置切割 DNA,会在识别序列外切割出任意的粘性末端,因此可以定制切割序列。
克隆过程如下:在目的基因切割位点外设计IIS型限制酶识别位点酶切后该识别位点被消除,不会出现在插入片段中,因此插入片段与载体连接后不会被二次切割;载体上含有与目的基因的切割位点互补的粘性末端,可以与之进行连接且不会引入新的序列,从而实现无缝克隆。
基于上述原理的 Golden Gate Assembly Kit(BsmBl),包含酶切连接所需要的酶,且以 Mix形式出现,加样更便捷,单次反应最高可进行 16个片段的连接,充分满足各种实验需求。
实验原理
注意:
1. 此处的载体通过酶切得到,需选择带有BsmBI酶切位点的载体。除此之外,载体还可以通过PCR获得,引物设计见下文。
2. 插入片段通过PCR方式获得,需将酶切位点通过引物加入片段末端。推荐使用高保真酶(REF: T1211)扩增,保证扩增产物正确性。
3. 红色的“TTTTTT”为保护碱基示意,可根据不同酶进行调整,推荐使用6个。
4. 上图仅展示了单片段的连接过程,更多片段的连接原理与之一致,只需改变粘性末端的序列即可增加连接片段数。
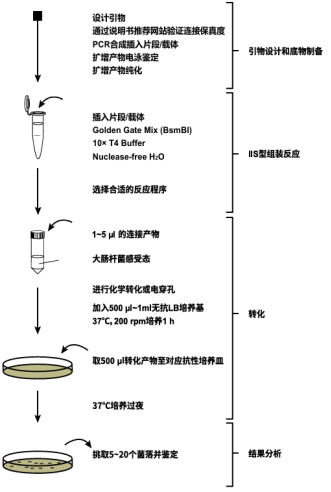
实验流程
引物设计指南
通过 PCR引入BsmBI的识别序列,识别序列加在引物的5'端,为确保限制酶能稳定结合到 DNA 双链上并发挥切割作用,需在识别序列末端加上保护碱基。保护碱基的数量和种类不固定(具体可查看兰博利德《快速内切酶使用指南》),推荐保护碱基为6bp,可以保证大部分普通酶切。由于切割位点在识别序列下游且可以是任意序列,因此有2种常见的引物设计方法,如下图所示:
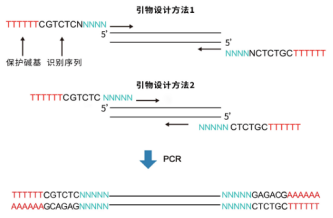
引物设计方法1:
此种引物设计方法为保护碱基与酶切位点即“TTTTTTCGTCTCNNNNN”均通过引物引入,其他序列则为插入片段上的序列(且必须大于15 bp),两部分共同构成引物。
引物设计方法2:
此种引物设计方法为保护碱基与识别序列即“TTTTTTCGTCTCN”通过引物引入,而“NNNN”则是插入片段的序列。
注 1:两种引物设计主要区别在于“NNNN”是否是插入片段中的序列。为保证无缝克隆,若插入片段与载体均通过 PCR 获得,则“NNNN”必须是片段或者载体(二选一)中的序列,即片段与载体需用两种不同的引物设计方式获得。
注 2:用于连接的粘性末端“NNNN”的序列对连接特异性存在较大影响,虽然理论上存在 256 种组合,但需排除回文序列。推荐使用以下网址中的工具进行粘性末端设计:https//goldengate.neb.com/#!/。
此外,引物设计还需注意以下几点:
1. 为保证扩增正确性,通常要求扩增产物<5kb,以此为前提设计引物;
2. 与模板配对的引物部分 Tm 值应在 58~60°C之间,此时扩增效果最好;
3. 引物内和引物间不能包含互补序列,以避免发夹形成;
4. 引物的质量对后续连接的影响巨大,即使连接所需的粘性末端只有一个碱基发生突变,也可能导致连接失败,建议选择可靠的基因合成公司。
PCR 注意事项
1. 建议针对每个片段优化 PCR条件,以确保单一的PCR产物;
2. 若 PCR 获得多个条带,必须凝胶纯化目标 DNA。否则将导致组装失败或克隆效率大大降低。胶回收 DNA时,尽量减少紫外切胶的时间,或选择蓝光切胶,以减少紫外线对 DNA的损伤;
3. 长片段 DNA(>5 kb)在胶回收时更容易被损伤,因此推荐使用多个小片段进行连接,而不是单个大片段;
4. 使用 ddHzO或10 mM Tris 缓冲液(pH 8.0)洗脱 DNA,避免使用 TE 缓冲液洗脱 DNA,以防止对下游连接产生抑制;
5. PCR 产物可以未经纯化直接用于连接反应,但一般只限于单片段克隆。多片段的连接仍需对 PCR产物进行纯化。
预克隆指南
预克隆即将 PCR 获得的 DNA片段插入到一个中间载体(预克隆供体载体,通常在3kb左右),然后将其与其他插入片段一同加入Golden Gate 体系中完成最终组装。
预克隆一般通过 TOPO克隆完成,将PCR产物通过拓扑异构酶连接到 TOPO 载体上,载体含有选择基因供我们筛选阳性克隆。预克隆方法并不固定,可根据您的习惯自行选择克隆手段。
以下以 TOPO 克隆进行预克隆流程示例:
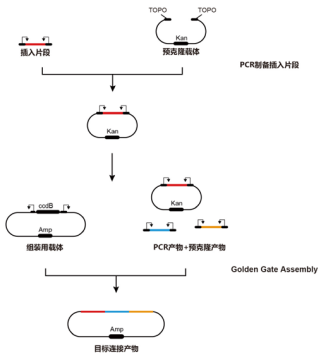
注 1:片段与载体中的黑色箭头代表限制酶酶切方向,从识别位点指向切割位点。
注 2:ccdB 是一种大肠杆菌致死基因,无外源基因插入时致死,插入片段可破坏ccdB 基因的表达,从而在转化时仅允许含插入片段的菌落生长。对于超过 80% 一致性的重复/同源序列,推荐使用预克隆;当插入片段数>5时,建议使用预克隆分步克隆至目标载体。
实验步骤
1、于冰上配置以下反应体系:
组分 | 连接反应 | 负对照a |
载体 | 0.05 pmolb | 0.05 pmol |
片段 | 0.1 pmolc | - |
10× T4 DNA Ligase Buffer | 2μl | 2μl |
Golden Gate Mix (BsmBI) | 1μl | 1μl |
Nuclease-free H2O | To 20 μl | To 20 μl |
a. Golden Gate Assembly 通常不要求负对照。如果需要,可设置不加插入片段的反应体系为负对照。
b. 对于2000 bp的载体,0.05 pmol 用量为 60 ng,其他长度可根据这个比例自行计算;
c. 摩尔比为片段:载体 =2:1,所有插入片段之间比例为 1:1;当片段大于载体时二者用量互换。
注:片段越多,连接效率和阳性率越低;片段或载体过长,连接效率也会下降。按上述体系配制完成后,涡旋混匀,于PCR仪中进行反应,反应程序根据片段数按下文推荐程序选择。
2、推荐的反应程序
插入片段数 | 反应程序 |
1 | 42℃,5 min→65℃,5 min |
2~4 | 42℃,1 h→65℃,5 min |
5~10 | (42℃,1 min→22℃,1 min) ×30~60→65℃,5 min |
注:对于10个以上的片段组装,虽可实现单次成功组装目标片段,但阳性率和菌落数会出现显著下降,为保证实验成功率,对于10+的组装场景,建议分次组装。
可选恒温程序:
插入片段数 | 反应程序 |
1 | 30℃,5 min→65℃,5 min |
2~10 | 30℃,1~2 h→65℃,5 min |
注:恒温反应对于简单场景如单片段反应效率无影响,对于多片段则会随片段增多而出现一定程度效率下降,但对于2~5片段仍可维持70%以上效率,在条件允许时,建议优先使用变温程序。
3、反应结束后,可以直接用于转化,或者-20°保存备用。
4、重组产物转化
取 5~10 μL反应液,加入到100 μL感受态细胞中,缓慢吸打混匀,冰上放置30 min。42°C热激45~60sec,冰浴5 min。加500μl SOC或LB 培养基,37°C振荡培养40~60min(200rpm)。将菌液均匀涂布在含有对应抗生素的平板上,倒置于37°C过夜培养。
注 1:不同感受态细胞最后的克隆阳性率会有所差别,推荐使用转化效率>10° CFU/ug 的感受态细胞;
注 2:菌落数取决于 PCR 产物与线性化载体的数量和纯度;
注 3:阳性对照平板通常生长大量白色单菌落,阴性对照平板只生长很少的菌落。
5、阳性克隆检测
克隆完成后,需对产物进行筛选鉴定,一般有酶切与PCR两种方法。
酶切:
挑取 5~20 个菌落至1 ml对应抗性的 LB 培养基中过夜培养:第二天提取质粒,选择合适的限制酶进行酶切,并通过琼脂糖电泳对酶切结果进行分析,筛选得到目标质粒。
对于常规片段连接,5~10个菌落已可以筛选到目标产物,而重复/同源序列或超过 10片段的连接,则需要20个菌落以筛选正确连接产物。
PCR:
(1)设计合适的检测引物,根据插入片段长度灵活设计扩增产物长度。对于单片段插入,一般要求扩增长度在500bp~2kb之间,正向引物与反向引物分别设计在载体与片段上。对于多片段插入,则正反向引物均设计在载体上,扩增插入片段全长。
(2)挑取 5~20 个菌落至1 ml对应抗性的LB 培养基中过夜培养,将培养产物分别取1M至PCR体系中(30IPCR体系即可满足扩增需求)0
(3)扩增程序在常规 PCR 基础上将第一步变性由 95°C 3 min 延长至 95°C 10 min,其余步骤保持不变。扩增完成后进行琼脂糖电泳检测扩增产物是否正确。
(4)选择扩增正确的菌液进行质粒提取,获得目标连接产物。(推荐对正确的连接产物进行进一步的测序验证,防止PCR扩增时出现错误)。





